DMD基因疗法坎坷上市, 我们专访了王亚宁 第一现场
2024-07-23 15:16:41

8年前,Janet Woodcock力排众议批准第一个DMD药物eteplirsen;8年后,Peter Marks在诸多阻力下批准第一个DMD基因疗法Elevidys。
在接下来几年里,FDA生物制品评估和研究中心(CBER)将大力推动使用替代终点去加速批准药物。CBER主任Peter Marks不止一次在公开场合表达了这样的观点。他在华盛顿一个研讨会上说道:“如果不采取加速审批,很多患者将无法及时接受治疗,加速审批将成为许多基因疗法获得初步批准的常态。”接受Endpoints采访时也表示,FDA计划鼓励开发商使用加速批准途径,特别是针对罕见疾病的基因和细胞疗法。
Peter Marks的观点,非常直观地在首个杜氏肌营养不良症(DMD)基因疗法Elevidys获批历程中得到印证。

2023年6月,Elevidys以微抗肌萎缩蛋白(micro-dystrophin)作为替代终点加速批准上市。“我觉得这是开辟了CBER的一个新纪元。”前FDA药物审评和研究中心(CDER)定量药理学审评部部长、上海瑞宁康生物医药创始人兼CEO王亚宁博士这样认为。

王亚宁
王亚宁表示,FDA列出的审评证据中,由定量药理部门提供的一张分析图,合理解释了在4~5岁患者中,替代终点微抗肌萎缩蛋白水平与临床终点NSAA(北极星动态评估)数据呈正相关性。同时他也指出,采用类似微抗肌萎缩蛋白的生物标志物作为替代终点,可能面临一个难题,即这些生物标志物增加到什么阈值才具备临床获益。这也是当初aducanumab审评的核心。
拓展阅读
王亚宁:希望能消除对aducanumab审批的误解
“例如Elevidys的微抗肌萎缩蛋白水平,用药组相对安慰剂组是增加了,但增加多少程度才有临床意义,其实并没有那么清晰。但目前从微抗肌萎缩蛋白水平与NSAA的正相关关系,结合3期验证性临床的二级终点数据,可以看到Elevidys的微抗肌萎缩蛋白水平已经足够。推演至其他基因疗法上,我觉得将来会面临同样的问题。”
决定性证据出现
研发客:Elevidys在2023年6月获得加速批准之前,经历怎样的审评之路?
王亚宁:
最初,Elevidys开展的一项SRP-9001-102(102研究)随机、双盲、安慰剂对照的2期研究,探索4~7岁患儿的疗效和安全性。但试验在2021年7月宣布未达NSAA的主要临床终点。2期试验的失败使该疗法的上市进展延缓了约一年。
当时我已离开了FDA,但仍然记得是Peter Marks和另外一位CDER的senior official,两位以书面形式鼓励企业,尽快将数据交上来,甚至可以考虑加速批准。这表示,在2022年11月Sarepta公司递交的BLA申请被受理之前,FDA就已经有了鼓励的方向。
企业递交上市申请后,在FDA内部遇到很大阻力。以往DMD获批药物都是CDER审评,Elevidys是CBER首个审评的DMD药物。一开始FDA并不打算举行外部专家会议,但由于内部审评团队不支持,Peter Marks提出开专家会,通过外部专家讨论,最终结果是8:6支持批准。
研发客:FDA为什么支持加速批准Elevidys?
王亚宁:
Elevidys的加速批准是基于骨骼肌中微抗肌萎缩蛋白水平的增加,以此作为替代终点。而且,批准的是4~5岁亚组,6~7岁亚组没有批准。
Sarepta将102研究按年龄拆分出来单独统计,发现4~5岁组Elevidys与安慰剂的NSAA分差为+2.5,而6~7岁组的NSAA分差为-0.7。从数据表面看,真正看到效果的是4~5岁组。
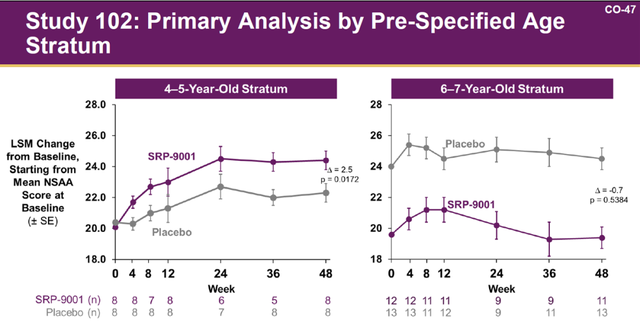
来源|FDA官网
Sarepta在专家会上合理地解释了为什么6~7岁组没有显示出疗效。6~7岁组在被分为用药组和安慰剂组时,基线分配发生了偏倚,部分基线症状较好的患者被分到安慰剂组,而基线症状较差的分到了用药组,从基线考虑这两组不具备可比性。同时,Sarepta也提出了解决办法,引用其他研究中的历史数据佐证了6~7岁亚组的疗效,来校正这种随机没做好的问题。
更重要的是,利用微抗肌萎缩蛋白作为替代终点,需要证明替代终点与临床终点呈现相关性。这其中,定量药理发挥了关键作用。
由于FDA内部审评团队的争议,Peter Marks撰写了独立的备忘录解释批准原因,其中由定量药理部提供的分析(见下图),合理解释了微抗肌萎缩蛋白水平与临床终点NSAA数据呈正相关性。我认为这张图是支持Elevidys获得加速批准的重要依据。
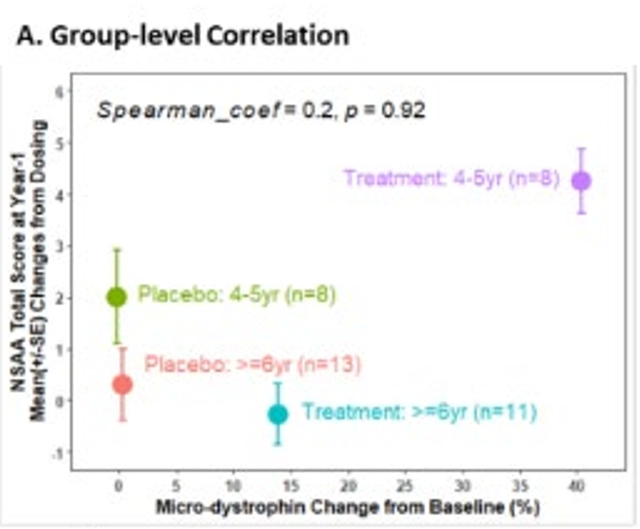
来源|FDA官网
这张图沿用了aducanumab同样的分析方法。其中两个点(紫色、绿色)是4~5岁组,另两个点(蓝色、红色)是6~7岁组,X轴是替代终点微抗肌萎缩蛋白,Y轴是NSAA评分,即主要临床终点。
分析结果与102研究结果一致,4~5岁的微抗肌萎缩蛋白水平越高,临床终点获益越大,呈正向结果。但6~7岁组却呈现了反向结果。沿用Sarepta的解释,6~7岁组在随机分配时发生了偏倚,需要将6~7岁组的数据进行校正至可比的程度,再重新进行分析,我认为会呈现一个正向结果。
次要终点支持完全批准
富有戏剧性的是,在2023年6月批准Elevidys加速上市之后(4~5岁亚组),其上市后验证性临床试验EMBARK(SRP-9001-301),2023年10月揭盲后再次失败,未到达主要临床终点NSAA评分。
“去年3期验证性临床失败后,很多华尔街投资机构来咨询我FDA会不会从市场上撤掉Elevidys。我看完具体数据后的答复是,FDA不仅不会撤掉Elevidys,还会把加速批准转正为完全批准,而且还会去掉年龄限制,从加速批准4~5岁亚组,拓展到6~7岁亚组。”王亚宁说。
如今Elevidys获得完全批准,也印证了王亚宁当初的观点。EMBARK试验中,两项关键次要终点Time to rise(TTR)、10米步行/跑步测试(10MWR),以及其他的次要终点SV95C、100MWR、Ascend 4,都均获得稳健且具有统计意义或者接近统计意义的提升,且在所有年龄段都如此,从而成为Elevidys获得完全批准、提供临床治疗获益的证据。
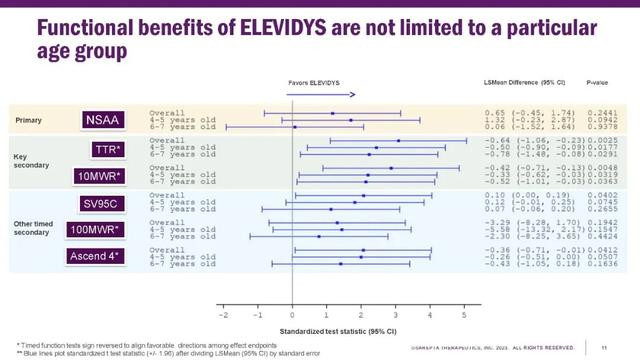

来源|Sarepta公司官网

关于主要临床终点NSAA评分差异未达到统计学意义(P值为0.2441)的结果,王亚宁表示,用药组相比安慰剂组有改善趋势,两组分别提高2.6分和1.9分,只是没有达到统计学意义。“熟悉统计的人都知道,P值是样本量的游戏,如果样本量足够大,观测到的用药组和安慰剂组之间的差别是可以得到P值小于0.05的。102和EMBARK两个试验都观察到了用药组优于安慰剂组的趋势,而且还有二级终点那么多阳性的结果,如果药物跟安慰剂没有差别,观察到这样结果的概率是很低的。”
值得关注的是,此次FDA除了授予可行走的DMD患者完全批准外,还授予不可行走患者以加速批准。
对此FDA给出的解释是,结合可行走患者中的临床数据,102研究显示4~5岁患儿微抗肌萎缩蛋白水平与NSAA呈相关性,EMBARK显示微抗肌萎缩蛋白水平与TTR、10MWR和Ascend 4呈相关性,再结合SRP-9001-103队列3中,不可行走患者在探索性临床终点上肢表现测试(PUL)的积极结果、以及对微抗肌萎缩蛋白水平的影响,FDA认为微抗肌萎缩蛋白水平的增加可以合理预测不可行走人群的临床获益。
同时,FDA要求Elevidys在另一项验证性临床研究ENVISION(SRP-9001-303)中,验证不可行走和年龄较大的可行走DMD患者的疗效,目前研究正在进行中。
研发客:EMBARK试验在临床终点选择上给我们带来哪些启示?
王亚宁:
临床终点选择是一个学习的过程。在一个没有太多经验的疾病领域,例如DMD,什么样的临床终点足够敏感、同时有足够的临床意义,体现出产品的真实疗效,企业和FDA都是在摸索中不断学习。
如果一开始选择的临床终点在实践中不够敏感,可以参考Elevidys,经历了102研究、EMBARK试验几次失败后,考虑不选择现有的NSAA作为主要临床终点,而选择那些二级终点,或者其他具有临床意义也更敏感的终点,进一步实践是否适合作为主要终点。
以往也遇到过类似案例。例如红斑狼疮药物belimumab,第一项2期临床试验失败后,企业对临床终点进行了优化,找到更加敏感且具有临床意义的新终点,并得到了FDA的同意,接下来的两个3期试验中以新临床终点为主要终点,最终试验成功获得批准,而当初不敏感的终点在两个3期试验中再次显示出不敏感性。早年一个叫做LJP的小公司,就是因为从头到尾始终用同样的这个不敏感临床终点研究一个红斑狼疮新药,而最终在完成一个2期、三个3期后以失败告终。
研发客:将来定量药理在基因疗法审评中,是不是可以发挥更多作用?
王亚宁:
传统意义上,定量药理大多用于证明新药疗效,剂量优化平衡获益风险比,为精准医疗的个体化用药提供依据,偶尔也可以豁免临床试验,但从阿尔茨海默病药物aducanumab的批准开始,至ALS药物tofersen和Elevidys的加速批准,定量药理都发挥了关键性作用,建立一个全新的替代终点而使得加速批准成为可能。例如需要当证明或量化某些基因治疗产品表达的蛋白或多肽作为替代终点、与临床终点获益之间呈相关性时,定量药理可以发挥重要的作用。
拓展阅读
从FDA幕后走向台前,定量药理经历了什么?
因为基因治疗的治病机理非常清晰,就是去修正或补偿那些缺陷或变异的基因,表达出正常蛋白或阻断异常蛋白。例如Elevidys治疗DMD,能证明替代终点微抗肌萎缩蛋白与临床终点NSAA呈线性相关,就表示微抗肌萎缩蛋白降低到足够程度就可以作为替代终点来支持加速批准。
所以我认为,将来FDA想要用替代终点支持更多的基因疗法上市时,定量药理将成为重要的分析工具。
倾听患者声音≠降低审评标准
研发客:Elevidys的完全批准,会不会有FDA降低审评标准之嫌?
王亚宁:
我觉得不需要担心这个问题,因为这是第一个DMD基因治疗药物,患者几乎处于无药可治状态,而当一个领域的药物多了之后,水涨船高,患者的治疗需求得到一定程度满足时,他们对风险获益平衡的评价也会随之改变,而监管机构也会听取患者的声音对审评标准做出调整。
而事实上,倾听患者声音,FDA已经强调和实践了很多年。2012年就建立以患者为中心的药物研发(PFDD)计划,并通过法律形式如《处方药用户付费法案》(PDUFA)、将患者经验纳入药物的开发和审查,并出台PFDD指南系列收集患者经验数据。
对于Elevidys的审评,我认为是真正的考虑了患者声音。在外部专家咨询会上,很多患者或者患者家属都表示在DMD这样的疾病面前他们愿意接受更多的风险和不确定性。我记得有一位患者这样和统计师辩论,统计师提出有假阳性概率的可能性,患者表示,如果一个药物有效,哪怕是5%的概率我也愿意尝试,但是如果不批准,我获益的概率就是0,我只需要一个大于0的概率就够了。
这足见患者们在疾病的折磨下对任何有希望的新药的渴望,他们不希望FDA剥夺他们作为使用者去尝试有风险但也可能有效的新药的权利。当初FDA肿瘤卓越中心主任Rick Pazdur作为FDA内部15个专家中唯一的临床专家支持aducanumab加速批准,他主要出发点也是患者的声音。
还有,与Elevidys类似,2016年,时任CDER主任Janet Woodcock,推翻了FDA所有审评团队以及外部专家组的意见,批准了首个DMD治疗药物eteplirsen。Janet Woodcock在退休后接受Endpoints采访时说,如果再遇到eteplirsen,还是会做同样的决定。由此可以看出,FDA高层的审评理念和决策是有迹可循、一脉相承的。